研究者専用ページ




臨床試験とは
新しい治療を開発するためには「臨床試験」が必要です
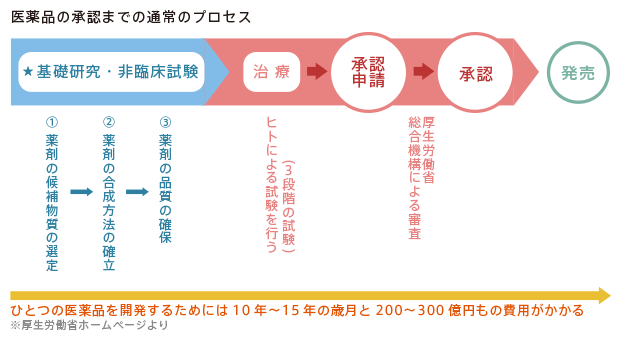
基礎研究や非臨床試験(上図の★の過程)が、はじめから小児がんをターゲットとして計画されることは非常に稀です。
現在小児がんに対して標準的に用いられている薬は、大部分が小児がんを特に意識して基礎研究や非臨床試験を行ったわけではなく、 成人用に開発された薬剤の有効性と安全性を小児がんで確認した結果、用いられているに過ぎません。
基礎研究と非臨床試験を経て開発された候補薬剤は、ヒトでのデータがなければ“くすり”とは呼べません。「臨床試験」とは、ヒト(患者または健常者)を対象として、薬剤の効果、安全性や体内での動きのデータを調べる目的で行うものです。 通常、以下の三つの段階に分けて考えられています。
1. 第Ⅰ相試験(用量の決定)
ある候補薬剤が初めてヒトに投与される際には、最初の何名かの患者さんは、安全と考えられるごく少量の薬剤を投与するような計画で試験が行われます。安全性を確認しながら少しずつ投与量を増やし、副作用が一定頻度以上に認められるようになったところで止め、集めたデータから推奨用量が決められます。がんの場合は、他に有効な薬のない再発がんや進行がんの患者さんが第Ⅰ相試験の対象となります。同じ薬剤が種々のがんの患者さんに 試される中で、どのようながんに対して第Ⅱ相試験を行うべきなのか見当をつける、という役割もあります。
2. 第Ⅱ相試験(有効性と安全性の検討)
第Ⅰ相試験で決定した推奨用量の薬剤を投与し、あらかじめ決められた評価項目を判定します。従来は、患者さんの20%以上に腫瘍の縮小が観察されることが有効性を測る基準でした。分子標的薬剤のような新しい薬剤では、腫瘍が縮小しなくても患者さんを延命できるケースが多々あるため、このような基準も変わりつつあります。この段階で有効性と安全性が確認されれば、厚生労働省によって、特定のがんに対する治療薬として製造販売が承認される可能性が出てきます。
3. 第Ⅲ相試験(従来の標準治療との比較)
通常は、患者さんを試験に登録するときに、標準的治療を受けるグループと新規治療を受けるグループに無作為に振り分け、予定された数の患者さんが治療を受け、データが得られた後に、グループ間で結果を比較して優劣をつけます。近年、抗がん剤は単独ではなく、いくつかを併用して治療を 行うことがほとんどなので、薬剤同士を1対1で比較することは稀で、「標準治療」VS「標準治療+新薬」という比較を行うことがほとんどです。第Ⅲ相試験において有効であると判断された治療が“新しい標準治療”として学会発表や学術論文で認められると、専門医に広く用いられるようになり、「医学常識が変わる」ことになります。
通常は、患者さんを試験に登録するときに、標準的治療を受けるグループと新規治療を受けるグループに無作為に振り分け、予定された数の患者第Ⅲ相試験は通常、多くの患者さんの参加が必要で、観察期間も長期にわたって必要なため、ひとつの試験に数年以上費やされることもしばしばです。
「治験」と「臨床試験」の違いとは?
人での有効性や安全性について調べるのが「臨床試験」です。
「臨床試験」のうち、「新薬の承認申請を目的とした特殊な臨床試験」のことを特に「治験」といいます。「治験」を実施するためには、薬事法や関連法規の下、厳密なルールが定められており、たくさんの人的資源と予算がかかりますので、通常は製薬会社が自社の薬剤を商品として販売するために「治験」を行っています。
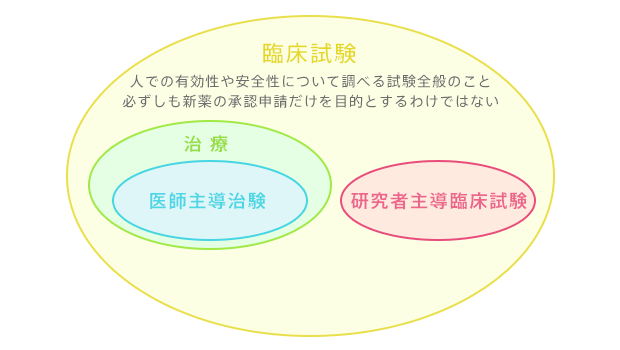
平成15年に薬事法が改正され、製薬会社でなくても医師や歯科医師が治験を行う事ができるようになりました。このような治験を「医師主導治験」と呼びます。ところが、厳密なルールや限られた予算の問題から、医師主導治験を行えるのは一部の限られた研究者となっているのが現状です。
一方で、医学研究者は、「既に承認された薬剤」の併用療法を工夫して治療成績を改善するための「臨床試験」を数多く行っており、これは「医師主導治験」と区別するために「研究者主導臨床試験」と呼ばれています。こうした臨床試験は、上述の第Ⅲ相試験に当たるものが多く、しばしば医学の常識を変え、その発展に貢献しています。
小児がんの分野においてはそれぞれの病気の患者数がきわめて少ないため、我が国だけで第Ⅲ相試験を行うことは困難です。このためSUCCESSでは、医学常識を変えるような大規模な臨床試験は欧米に任せ、我が国では第Ⅰ相試験や第Ⅱ相試験を数多く行って、小児がんに使える薬剤を増やすことに貢献すべきと考えています。このような理由から、SUCCESS治療開発支援センターでは医学研究者が臨床試験を行うために必要なデータ登録や解析などの業務を受託し、事務支援を行っています。